Indications:
⚫ is used to reduce the risk of stroke and systemic embolism in patients with non valvular atrial fibrillation.
⚫ is indicated for the treatment of deep vein thrombosis, which may lead to pulmonary embolism in patients undergoing knee or hip replacement surgery.
is indicated for the treatment of Pulmonary Embolism (P.E.).
⚫ is indicated
WARNING:
A- Premature discontinuation of Rivaroxaban increases the risk of thrombotic events: Premature discontinuation of any oral anticoagulant, including Rivaroxaban, increases the risk of thrombotic events. If anticoagulation with Rivaroxaban is discontinued for a reason other than pathological bleeding or completion of a course of therapy, consider coverage with another anticoagulant.
B- Spinal / Epidural Hematoma:
Epidural or spinal hematomas have occurred in patients treated with Rivaroxaban who are receiving neuraxial anesthesia or undergoing spinal puncture, these hematomas may result in long-term or permanent paralysis. Consider these risks when scheduling patients for spinal procedures. Factors that can increase the risk of developing epidural or spinal hematomas in these patients include:
1- Use of indwelling epidural catheters.
2- Concomitant use of other drugs that affect hemostasis, such as non-steroidal anti-inflammatory drugs (NSAIDs), platelet inhibitors, other anticoagulants.
3- A history of traumatic or repeated epidural or spinal punctures.
4- A history of spinal deformity or spinal surgery.
5- Optimal timing between the administration of Rivaroxaban and neuraxial procedures is not known. Monitor patients frequently for signs and symptoms of neurological impairment. If neurological compromise is noted, urgent treatment is necessary.
Consider the benefits and risks before neuraxial intervention in patients anticoagulated or to be anticoagulated for thromboprophylaxis.
Composition:Xarivan F.C.T Contain 2.5 or 10,or15or20 mg rivaroxaban.
Excipients:
Core:Croscarmellose
sodium,hypromellose, lactose
stearate,microcrystalline cellulose and sodium lauryl sulfate.
monohydrate,magnesium
Film:hypromellose,polyethyleneglycol,titanium dioxide,red iron oxide,yellow iron oxide. Mechanism of Action:
Rivaroxaban is a selective inhibitor of factor Xa; it is a selective inhibitor of FXa. It does not require a cofactor (such as Anti-thrombin III) for activity. Rivaroxaban inhibits free FXa and prothrombinase activity. Rivaroxaban has no direct effect on platelet aggregation, but indirectly inhibits platelet aggregation induced by thrombin. By inhibiting FXa, rivaroxaban decreases thrombin generation. Pharmacokinetics:
Absorption:
The absolute bioavailability of Rivaroxaban is dose-dependent, for the 10 mg dose, it is estimated to be 80% to 100% and is not affected by food; the absolute bioavailability of Rivaroxaban at a dose of 20 mg in the fasted state is approximately 66%. Coadministration of Rivaroxaban with food increases the bioavailability of the 20 mg dose (mean AUC and Cmax increasing by 39% and 76% respectively with food), Rivaroxaban 15 mg and 20 mg tablets should be taken with the meal, the maximum concentrations (Cmax) of Rivaroxaban appear 2 to 4 hours after tablet intake.
Distribution:
Plasma protein binding of Rivaroxaban is approximately 92% to 95%.
Excretion:
Following oral administration of Rivaroxaban dose, 66% of the dose was recovered in urine (36% as unchanged drug) and 7% is excreted by feces (as unchanged drug). The terminal elimination half- life of rivaroxaban is 5 to 9 hours in healthy subjects aged 20 to 45 years.
for reduction in the risk of recurrence of deep vein thrombosis and pulmonary embolism following initial 6 months treatment for DVT and/or PE.
⚫ is indicated for the prophylaxis of Deep Vein Thrombosis (DVT) following Hip or Knee
replacement surgery, which may lead to Pulmonary Embolism (PE).
Reduction of Risk of Major Cardiovascular Events in Patients with Chronic Coronary Artery Disease (CAD) or Peripheral Artery Disease (PAD) (for 2.5 mg
strength).
Contraindications:
is contraindicated in patients with:
- Active pathological bleeding.
- Severe hypersensitivity reactions to the drug or its components.
Adverse reactions:
- Increased risk of stroke after discontinuation in nonvalvular atrial fibrillation.
- Bleeding risk.
- Spinal / Epidural Hematoma.
Warnings and Precautions:
Increased risk of thrombotic events after Premature Discontinuation:
Premature discontinuation of any oral anticoagulant, including Rivaroxaban, increases the risk of thrombotic events. If anticoagulation with Rivaroxaban is discontinued for a reason other than pathological bleeding or completion of a course of therapy, consider coverage with another anticoagulant.
Risk of Bleeding:
Rivaroxaban increases the risk of bleeding and can cause serious or fatal bleeding. In deciding whether to prescribe Rivaroxaban to patients at increased risk of bleeding, the risk of thrombotic events should be weighed against the risk of bleeding. Promptly evaluate any signs or symptoms of need for blood replacement. Discontinue Rivaroxaban in patients with and consider the blood loss active pathological hemorrhage.
Spinal / Epidural Anesthesia or Puncture:
When neuraxial anesthesia (Spinal / Epidural anesthesia) or Spinal Puncture is employed, patients treated with anticoagulant agents for prevention of thromboembolic complications are at risk of developing an epidural or spinal hematoma which can result in long-term or permanent paralysis. An epidural catheter should not be removed earlier than 18 hours in young patients aged 20 to 45 years and 26 hours in elderly patients aged 60 to 76 years after the last administration of Rivaroxaban. The next Rivaroxaban dose is not to be administered earlier than 6 hours after the removal of the catheter. If traumatic puncture occurs, the administration of Rivaroxaban is to be delayed for 24 hours.
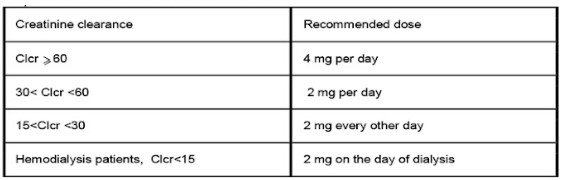
Use in Patients with Renal Impairment:
– Nonvalvular Atrial Fibrillation:
Periodically assess renal function as clinically indicated (i.e., more frequently in situations in which renal function may decline) and adjust therapy accordingly. Discontinue Rivaroxaban in patients who develop acute renal failure while on Rivaroxaban.
– Treatment of Deep Vein Thrombosis (DVT), Pulmonary Embolism (PE), and Reduction in the Risk of Recurrence of DVT and of PE:
Avoid the use of Rivaroxaban in patients with CrCl <30 mL/min due to an expected increase in Rivaroxaban exposure and pharmacodynamic effects in this patient population.
Prophylaxis of Deep Vein Thrombosis Following Hip or Knee Replacement Surgery:
Avoid the use of Rivaroxaban in patients with CrCl <30 mL/min due to an expected increase in Rivaroxaban exposure and pharmacodynamic effects in this patient population. Patients who develop acute renal failure while on Rivaroxaban should discontinue the treatment.
Use in Patients with Hepatic Impairment:
No clinical data are available for patients with severe hepatic impairment.
Avoid use of Rivaroxaban in patients with moderate and severe hepatic impairment or with any hepatic disease associated with coagulopathy.
Use with P-gp and Strong CYP3A4 Inhibitors or Inducers:
Avoid concomitant use of Rivaroxaban with combined P-gp and strong CYP3A4 inhibitors. Avoid concomitant use of Rivaroxaban with drugs that are combined P-gp and strong CYP3A4 inducers.
Patients with Prosthetic Heart Valves:
The safety and efficacy of Rivaroxaban have not been studied in patients with prosthetic heart valves. Therefore, use of Rivaroxaban is not recommended in these patients.
Acute PE in Hemodynamically Unstable Patients or Patients Who Require Thrombolysis or Pulmonary Embolectomy:
Initiation of Rivaroxaban is not recommended acutely as an alternative to unfractionated heparin in patients with pulmonary embolism who present with hemodynamic instability or who may receive thrombolysis or pulmonary embolectomy.
Pregnancy (category C):
Rivaroxaban should be used with caution in pregnant women and only if the potential benefit justifies the potential risk to the mother and fetus.
Nursing mothers:
It is not known if Rivaroxaban is excreted in human milk. A decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Geriatric:
Rivaroxaban in the elderly (65 years of age and older) was similar to that seen in patients younger than 65 years. Both thrombotic and bleeding event rates were higher in these older patients, but the risk-benefit profile was favorable in all age groups.
Pediatric Use:
Safety and effectiveness in pediatric patients have not been established.
Drug Interactions:
Drugs that Inhibit Cytochrome P450 3A4 Enzymes and Drug Transport Systems:
Interaction with Combined P-gp and Strong CYP3A4 Inhibitor: Avoid concomitant administration with known combined P-gp and strong CYP3A4 inhibitors (e.g., ketoconazole and ritonavir) Although clarithromycin is a combined P-gp and strong CYP3A4 inhibitor, pharmacokinetic data suggests that no precautions are necessary with concomitant administration with Rivaroxaban as the change in exposure is unlikely to affect the bleeding risk.
Interaction with Combined P-gp and Moderate CYP3A4 Inhibitors in Patients with Renal Impairment: Rivaroxaban should not be used in patients with CrCl 15 to <80 mL/min who are receiving
concomitant combined P-gp and moderate CYP3A4 inhibitors (e.g., erythromycin) unless the potential benefit justifies the potential risk.
Drugs that induce Cytochrome P450 3A4 Enzymes and Drug Transport Systems:
Avoid concomitant use of Rivaroxaban with drugs that are combined P-gp and strong CYP3A4 inducers (e.g., carbamazepine, phenytoin, rifampin, St. John’s wort).
Anticoagulants and NSAIDs/Aspirin:
Coadministration of enoxaparin, warfarin, aspirin, clopidogrel and chronic NSAID use may increase the risk of bleeding .Avoid concurrent use of Rivaroxaban with other anticoagulants due to increased bleeding risk unless benefit outweighs risk. Promptly evaluate any signs or symptoms of blood loss if patients are treated concomitantly
with aspirin, other platelet aggregation inhibitors, or NSAIDs.
Dosage and Administration:
Reduction in risk of stroke in Nonvalvular Atrial Fibrillation:
Patients with creatinine clearance (CrCl > 50 ml/min); one tablet of 20 mg once daily with the evening meal.
Patients with (CrCl 15 to 50 mL/min); one tablet of 15 mg once daily with the evening meal. Treatment of Deep Vein Thrombosis (DVT):
One tablet of 15 mg twice daily with food for first 21 days, after 21 days, transition to 20 mg once daily with food for remaining treatment.
Treatment of Pulmonary Embolism (PE):
One tablet of 15 mg twice daily with food for first 21 days, after 21 days, transition to 20 mg once daily with food for remaining treatment.
Reduction in the Risk of Recurrence of DVTand/or PE in patients at continued risk for DVTand/or PE
One tablet of 10 mg once daily with or without food, after at least 6 months of standard anticoagulant treatment. at pproximately the same time each day.
Prophylaxis of DVT following Hip or Knee replacement surgery:
– Hip replacement: one tablet of 10 mg once daily for 35 days, with or without food. The initial dose should be taken 6 to 10 hours after surgery provided that hemostasis has been established
– Knee replacement: one tablet of 10 mg once daily for 12 days, with or without food. The initial dose should be taken 6 to 10 hours after surgery provided that hemostasis has been established.
Reduction of Risk of Major Cardiovascular Events (CV Death, MI, and Stroke) in Chronic CAD or PAD:
No dose adjustment needed based on CrCl. 2.5 mg twice daily, plus aspirin (75-100 mg) once daily .Take with or without food.
Important food effect information:
The 15 mg and 20 mg tablets should be taken with food, while the 10 mg tablets can be taken with or without food, In the nonvalvular atrial fibrillation efficacy study it was taken with the evening meal. Switching to and from Rivaroxaban to other anticoagulant:
-Switching from warfarin to Rivaroxaban: When switching patients from warfarin to Rivaroxaban discontinue warfarin and start Rivaroxaban as soon as the International Normalized Ratio (INR) is below 3.0 to avoid periods of inadequate anticoagulation.
– Switching from Rivaroxaban to warfarin: No clinical trial data are available to guide converting patients from Rivaroxaban to Warfarin, and because Rivaroxaban affects (INR) so (INR) measurements made during coadministration with warfarin may not be useful for determining the appropriate dose of warfarin. One approach is to discontinue Rivaroxaban and begin both a parenteral anticoagulant and warfarin at the time the next dose of Rivaroxaban would have been taken.
-Switching from Rivaroxaban to anticoagulants other than warfarin: For patients currently taking Rivaroxaban and transitioning to an anticoagulant with rapid onset, discontinue Rivaroxaban and give the first dose of the other anticoagulant (oral or parenteral) at the time that the next Rivaroxaban dose would have been taken.
– Switching from anticoagulants other than warfarin to Rivaroxaban: For patients currently receiving an anticoagulant other than warfarin, start Rivaroxaban between (0 to 2 hours) prior to the next scheduled evening administration of the drug (e.g., low molecular weight heparin or non-warfarin oral anticoagulants) and omit administration of the other anticoagulant. For unfractionated heparin being administered by continuous infusion, stop the infusion and start Rivaroxaban at the same time. Discontinuation for surgery and other interventions:
If anticoagulation must be discontinued to reduce the risk of bleeding with surgical or other procedures, Rivaroxaban should be stopped at least 24 hours before the procedure to reduce the risk of bleeding. In deciding whether a procedure should be delayed until 24 hours after the last dose of Rivaroxaban, the increased risk of bleeding should be weighed against the urgency of intervention. Rivaroxaban should be restarted after the surgical or other procedures as soon as adequate hemostasis has been established, noting that the time to onset of therapeutic effect is short. If oral medication cannot be taken during or after surgical intervention, consider administering a parenteral anticoagulant.
Missed dose:
If a dose of Rivaroxaban is not taken at the scheduled time, administer the dose as soon as possible on the same day as follows:
– For patients receiving 15 mg twice daily: The patient should take Rivaroxaban immediately to ensure intake of 30 mg Rivaroxaban per day. In this particular instance, two 15 mg tablets may be taken at once. The patient should continue with the regular 15 mg twice daily intake as recommended on the following day.
– For patients receiving 20 mg, 15 mg or 10 mg once daily: The patient should take the missed Rivaroxaban dose immediately.
Administration options:
– For patients who are unable to swallow whole tablets, 10 mg, 15 mg or 20 mg Rivaroxaban tablets may be crushed and mixed with applesauce immediately prior to use and administered orally. After the administration of a crushed Rivaroxaban 15 mg or 20 mg tablet, the dose should be immediately followed by food.
– Administration via nasogastric (NG) tube or gastric feeding tube: After confirming gastric placement of the tube, 10 mg, 15 mg or 20 mg Rivaroxaban tablets may be crushed and suspended in 50 mL of water and administered via an NG tube or gastric feeding tube. Since Rivaroxaban absorption is dependent on the site of drug release, avoid administration of Rivaroxaban distal to the stomach which can result in reduced absorption and thereby, reduced drug exposure. After the administration of a crushed Rivaroxaban 15 mg or 20 mg tablet, the dose should then be immediately followed by enteral feeding.
Crushed 10 mg, 15 mg or 20 mg Rivaroxaban tablets are stable in water and in applesauce for up to 4 hours.
Overdosage:
Overdose of Rivaroxaban may lead to hemorrhage. Discontinue Rivaroxaban and initiate appropriate therapy if bleeding complications associated with Overdosage, had occurred. A specific antidote of Rivaroxaban is not available. Rivaroxaban systemic exposure is not further increased at single dose > 50 mg due to limited absorption. The use of activated charcoal to reduce absorption in case of Rivaroxaban overdose may be considered, due to the high plasma protein binding, Rivaroxaban is not expected to be dialyzable.
Storage Conditions: Store at room temperature,between (20-25)° C.
packaging: Xarivan (2.5,10,15,20) 2 blisters in carton box,each blister contains 10 F.C.T